analyse-rna-seq
View the Project on GitHub omics-school/analyse-rna-seq
Contenu :
L’objectif de cette partie est d’installer conda et les logiciels nécessaires à l’analyse RNA-seq des données O. tauri.
1.1 Installer conda
Conda est un gestionnaire de logiciels et d’environnements très utilisé en bioinformatique.
Miniconda est une distribution qui permet d’installer conda sous Windows, Mac OSX et Linux. Puisque nous travaillons sous Linux (sous Windows 10 certes, mais nous vous rappelons que WSL est un système Linux), nous allons installer la version Linux de Miniconda.
Sous Windows, ouvrez un terminal Ubuntu. Si vous avez oublié comment faire, consultez le tutoriel Unix.
Déplacez-vous ensuite dans votre répertoire utilisateur Unix :
$ cd
Vérifiez que la commande pwd renvoie bien /home/duo.
Téléchargez ensuite miniconda :
$ wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh
Installez miniconda :
$ bash Miniconda3-latest-Linux-x86_64.sh -b -f
Cette étape va prendre quelques secondes.
Initialisez ensuite conda :
$ ./miniconda3/bin/conda init
Attention, la commande débute par un point .
Fermez enfin votre terminal.
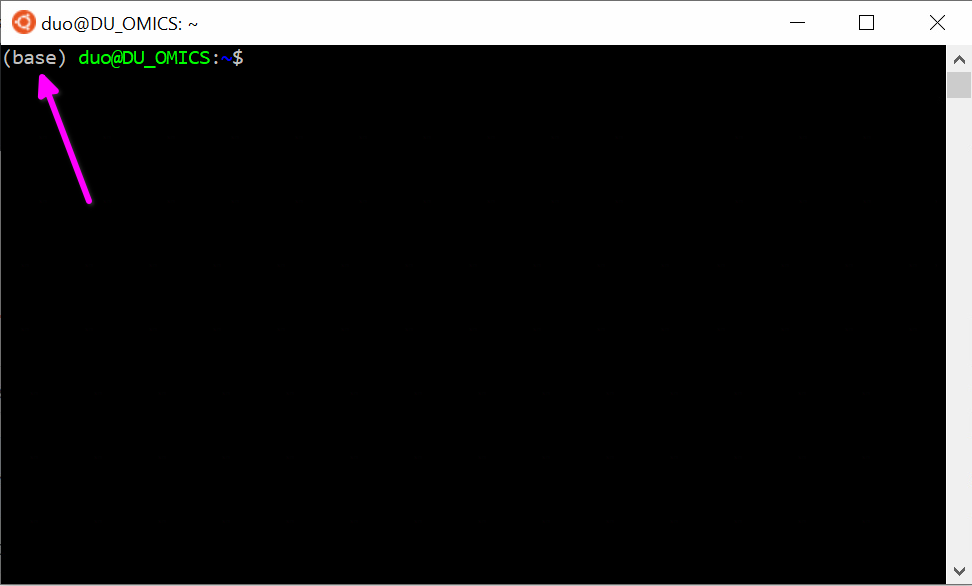
Relancez un nouveau terminal. Vous devriez voir (base) à gauche de votre invite de commande (comme ici).
{kind=link}
Vérifiez enfin que conda est fonctionnel en tapant la commande :
$ conda --version
Si vous obtenez conda 4.10.3 ou une version supérieure : bravo 🎉
1.2 Installer mamba
Conda est parfois lent à installer un environnement, c’est-à-dire à installer l’ensemble des outils nécessaires pour une tâche particulière, ici notre analyse RNA-seq.
Nous allons installer mamba qui accélerera l’installation des autres logiciels :
$ conda install mamba -n base -c conda-forge -y
1.3 Créer l’environnement rnaseq-env
Nous souhaitons maintenant installer tous les logiciels nécessaires à l’analyse RNA-seq. Nous pourrions le faire dans l’environnement par défaut de Miniconda (qui s’appelle base comme l’indique le (base) à gauche de votre invite de commande) mais ce serait une très mauvaise pratique.
Nous allons donc créer un environnement conda dédié à notre analyse RNA-seq. Sans grande originalité, nous appellerons cet environnement rnaseq-env :
$ conda create -n rnaseq-env -y
Une fois l’environnement créé, il faut l’activer, c’est-à-dire dire explicitement à conda que voulons l’utiliser :
$ conda activate rnaseq-env
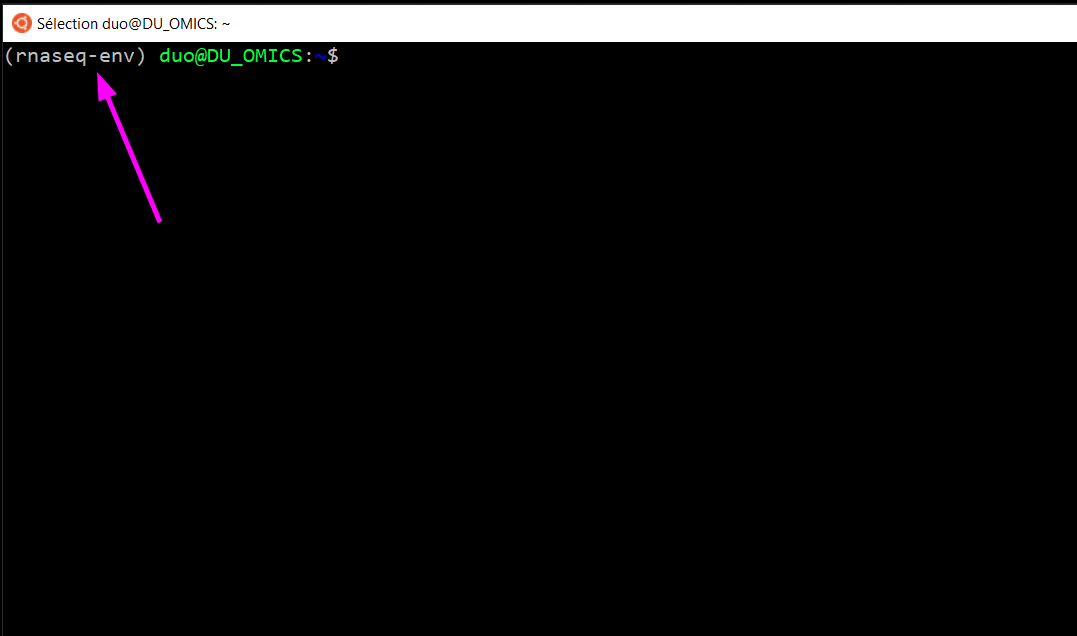
L’indication (base) à gauche de votre invite de commande est maintenant remplacé par (rnaseq-env) (comme ici).
{kind=link}
1.4 Installer les logiciels nécessaires
Voici la liste des logiciels dont nous avons besoin :
- SRA Toolkit pour télécharger les données de séquençage.
- FastQC pour le contrôle qualité des données de séquençage.
- Bowtie2 pour l’indexation du génome de référence puis l’alignement des reads sur le génome de référence.
- SAMtools pour la manipulation des fichiers d’alignements (conversion en binaire, tri et indexation).
- HTSeq pour le comptage du nombre de reads alignés sur chaque gène.
Installons ces logiciels dans l’environnement conda rnaseq-env que nous venons de créer. Pour accélérer l’installation, nous utilisons ici mamba :
$ mamba install -c conda-forge -c bioconda sra-tools fastqc bowtie2 samtools htseq -y
L’installation des logiciels et de leurs dépendances va prendre quelques minutes.
Pour terminer, vérifiez que les logiciels sont bien installés en affichant leurs versions :
Remarque : les versions peuvent légèrement différer.
$ fasterq-dump --version
"fasterq-dump" version 2.11.0
Remarque : fasterq-dump est un outil fourni par SRA Toolkit.
$ fastqc --version
FastQC v0.11.9
$ bowtie2 --version
/home/pierre/.soft/miniconda3/envs/rnaseq/bin/bowtie2-align-s version 2.4.4
64-bit
Built on default-51d38ee7-0be3-4ea2-a3d8-c7558b235b3c
Mon May 24 01:26:39 UTC 2021
[...]
$ samtools --version
samtools 1.14
Using htslib 1.14
Copyright (C) 2021 Genome Research Ltd.
[...]
$ htseq-count --version
1.99.2
Bravo ✨ Vous avez installé Miniconda, créé un environnement conda et installé tous les logiciels nécessaires pour votre analyse RNA-seq.
Vous pouvez passer à l’étape suivante : ➡️
Remarque : dans un environnement conda, on peut accéder rapidement aux versions des logicels installés avec la commande conda list.
L’utilisation de grep filtre ensuite le résultat. Par exemple
$ conda list | grep htseq
htseq 1.99.2 py39haf81c86_0 bioconda
ou
$ conda list | grep sra
sra-tools 2.11.0 pl5262h314213e_1 bioconda